

Você isolou uma bactéria com atividade antibiótica promissora. Ou encontrou um fungo que degrada poluentes de forma surpreendente. Ou identificou uma alga com potencial para biocombustíveis. A pergunta que não quer calar é: o que exatamente esse organismo tem no seu DNA que explica essas capacidades?
A resposta está no genoma completo.
E pela primeira vez na história, obter essa resposta deixou de ser privilégio de grandes centros de pesquisa. Hoje, com investimento acessível e tempo reduzido, qualquer pesquisador pode ter acesso ao manual de instruções completo do seu microrganismo de interesse.
Vamos entender exatamente o que é uma análise de genoma completo, como ela funciona tecnicamente, e o que você ganha com ela.

O que é o Genoma?
A biblioteca de instruções genéticas
O genoma é o conjunto completo de DNA de um organismo. É literalmente o manual que contém todas as instruções para construir e operar aquele ser vivo.
Para microrganismos:
- Bactérias: Genomas tipicamente entre 0,5 a 15 milhões de pares de bases (Mb)
- Fungos: Genomas entre 8 a 45 Mb, podendo chegar a mais de 100 Mb em alguns casos
- Algas: Altamente variáveis, de 12 Mb até 150 Mb dependendo da espécie
A diferença crucial: Enquanto o genoma humano possui cerca de 3.200 Mb com apenas ~20.000 genes codificadores de proteínas, uma bactéria de 4 Mb pode ter 4.000 genes – uma densidade muito maior. Isso significa que genomas microbianos são extremamente eficientes: quase não há “DNA supérfluo”.
Por que Genoma “completo”?
Existem diferentes níveis de análise genética:
1. Sequenciamento de genes marcadores(16S rRNA, ITS):
- Identifica o organismo
- Tem uma resolução de identificação de espécie limitada
- Não revela capacidades metabólicas
- Não mostra genes específicos
3. Genoma completo:
- Alta cobertura (30x ou mais significa cada base foi lida, em média, 30 vezes)
- Montagem de qualidade
- Anotação funcional completa
A analogia do quebra-cabeça:
O sequenciamento de genes marcadores é como pegar uma única peça de um quebra cabeça e tentar adivinhar qual é a imagem. Você pode conseguir descobrir que é uma praia, por conter partes de areia e água, mas não sabe exatamente qual a praia ou o contexto da imagem. Quando montamos o genoma completo, temos uma visão geral dessa imagem. Conseguimos identificar o Cristo Redentor ao fundo, saber se o dia está ensolarado ou nublado, e até mesmo uma boa estimativa de quantas pessoas estão na praia neste dia. Da mesma forma quando montamos um genoma completo, podemos identificar seus genes, saber exatamente qual é a espécie do organismo e ter uma boa ideia do potencial metabólico dele.

O que é a análise de Genoma completo?
A análise de genoma completo é um processo de múltiplas etapas que vai desde a extração do DNA até a interpretação biológica dos dados. Vamos detalhar cada etapa técnica.
Etapa 1: Preparação da amostra e extração de DNA
Ponto de partida:
- Cultura pura do microrganismo, ou
- DNA já purificado (mínimo ~1 µg)
Controle de qualidade essencial: A pureza do DNA é crítica. Utilizamos espectrofotometria para verificar:
- Razão 260/280: Deve estar entre 1,8-2,0 (indica pureza sem contaminação proteica)
- Razão 260/230: Deve ser >2,0 (indica ausência de contaminantes orgânicos)
- Concentração: Verificada por fluorimetria (Qubit) para precisão
Por que isso importa: DNA contaminado ou degradado resulta em sequenciamento de baixa qualidade, montagens fragmentadas e anotações imprecisas.
Etapa 2: Confirmação de identidade (Sequenciamento Sanger)
Antes de investir no sequenciamento completo, confirma-se a identidade do organismo através do sequenciamento Sanger de genes marcadores:
Para bactérias: Gene 16S rRNA (~1.500 pares de bases)
- Permite identificação até gênero ou espécie
- Comparação com banco de dados (NCBI, RDP, SILVA)
- Detecta possíveis contaminações
Para fungos: Região ITS (Internal Transcribed Spacer)
- Marcador universal para fungos
- Permite identificação até gênero ou espécie
- Essencial para validar pureza da cultura
Para algas: 18S rRNA ou outros marcadores específicos
O que isso previne: sequenciar o organismo errado ou detectar contaminação antes de investir no sequenciamento completo, economizando tempo e recursos.
Etapa 3: Preparação de biblioteca (Nextera)
O DNA genômico precisa ser fragmentado e preparado para sequenciamento. A tecnologia Nextera utiliza transposases – enzimas que simultaneamente fragmentam o DNA e adicionam adaptadores necessários para sequenciamento.
Vantagens da Nextera:
- Menor quantidade de DNA inicial necessária (~50 ng)
- Processo mais rápido (protocolo de poucas horas)
- Fragmentação mais uniforme
- Menor viés de cobertura
O processo técnico:
- Transposases fragmentam DNA em pedaços de ~300-800 pb
- Adaptadores são adicionados nas extremidades
- PCR amplifica os fragmentos
- Purificação e quantificação da biblioteca
- Normalização para concentração ideal
Etapa 4: Sequenciamento Illumina NextSeq
A plataforma Illumina NextSeq é o padrão-ouro atual para sequenciamento de genomas microbianos.
Especificações técnicas:
Paired-End (PE) 300 pb:
- Cada fragmento é lido de ambas as extremidades
- Leituras de 2×300 bases (totalizando 600 pb por fragmento)
- Permite melhor montagem e resolução de regiões repetitivas
Cobertura >30x:
- Cada posição do genoma é lida em média 30 vezes
- Maior confiança nas bases chamadas (>99,9% de acurácia)
- Permite identificar variantes e possível heterogeneidade
- Detecta erros de sequenciamento por consenso
Por que isso importa:
- 10x de cobertura: adequado para draft genomes
- 30x de cobertura: genomas de alta qualidade
- 50-100x de cobertura: detecção de variantes raras
Output típico:
- Bactéria de 5 Mb com 30x = ~150 Mb de dados (~250.000 reads)
- Fungo de 35 Mb com 30x = ~1.050 Mb de dados (1.750.000 reads)
Etapa 5: Análise Bioinformática – Controle de Qualidade
Antes de qualquer manipulação, submetemos os dados brutos ao FastQC, a ferramenta padrão para o controle de qualidade de sequenciamento NGS. Este processo funciona como uma inspeção técnica onde o software gera uma série de relatórios estatísticos que avaliam desde a distribuição da qualidade por base até a presença inesperada de sequências repetitivas. Através de métricas como o Phred Quality Score (Q), conseguimos visualizar se a corrida de sequenciamento entregou a precisão prometida. Se o FastQC aponta um sinal anormal em determinadas regiões das leituras, sabemos exatamente onde o pipeline precisa intervir. É este diagnóstico que orienta a etapa seguinte de limpeza, garantindo que apenas os dados que atingem um determinado padrão sigam para a análise.
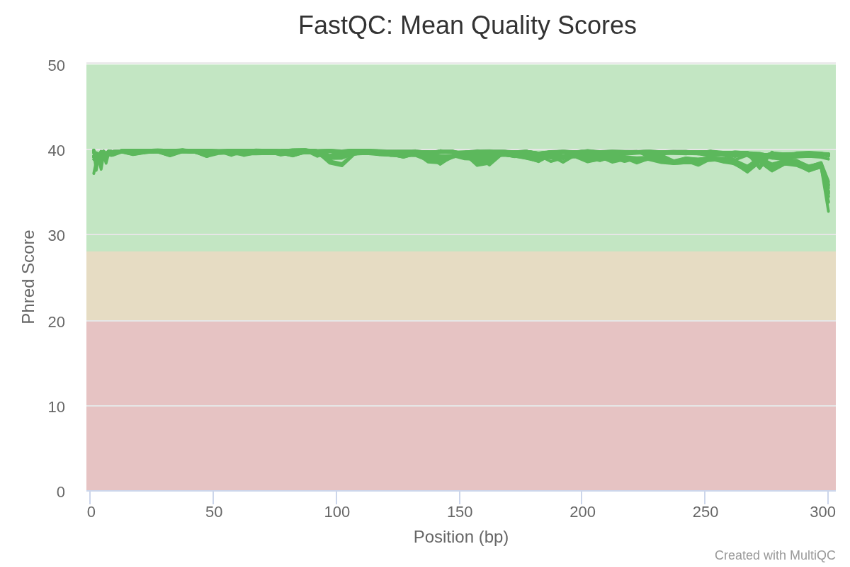
Após o sequenciamento, os dados brutos (arquivos FASTQ) contêm não apenas o código genético do seu microrganismo, mas também “ruídos” técnicos inevitáveis do processo. Imagine que você recebeu as páginas de um livro, mas algumas têm manchas de tinta nas bordas e outras contêm grampos que prendiam as folhas. A limpeza de dados, ou trimming, é a etapa onde removemos esses artefatos para garantir que a montagem final seja fiel à realidade biológica do genoma.
O que removemos e por quê?
- Adaptadores de sequenciamento: As sequências sintéticas utilizadas na etapa 3 para prender o DNA à flowcell do sequenciador. Se não forem removidas, o software de montagem tentará usa-las como partes reais do nosso quebra cabeça, incorporando essas sequências ao genoma, gerando erros.
- Bases de baixa qualidade: A precisão dos sequenciadores Illumina tende a cair ligeiramente no final dos fragmentos. Manter essas bases “duvidosas” aumenta o risco de identificar mutações inexistentes ou criar quebras artificiais na montagem.
Para garantir a qualidade dos dados usados, utilizamos o Trimmomatic, uma das ferramentas mais citadas da literatura científica. Ele opera através de uma “janela deslizante” (sliding window): o algoritmo percorre cada leitura de DNA utilizando uma janela de alguns nucleotídeos e, assim que a qualidade média desta janela cai abaixo de um limiar estatístico, a extremidade é cortada.
O resultado é um conjunto de dados limpos e de qualidade (Phred score >30, o que significa 99,9% de acurácia por base), prontos para serem montados no quebra-cabeça final. Sem esse cuidado, os dados brutos de má qualidade invariavelmente produzem resultados biológicos pouco confiáveis.

Etapa 6: Montagem do Genoma (Assembly)
Se o sequenciamento nos entrega milhões de pequenos fragmentos, a montagem é o momento de reconstruir o “texto” original do DNA. Para isso, existem dois caminhos metodológicos principais:
Montagem por referência: Utiliza-se um genoma já conhecido como “molde” para alinhar as novas peças. É eficiente para identificar pequenas mutações em linhagens bem estudadas, mas pode ignorar regiões totalmente novas ou rearranjos genômicos exclusivos da sua amostra.
Montagem de novo: As peças são unidas do zero, baseando-se apenas na sobreposição entre elas, sem a necessidade de um guia externo. É como montar um quebra-cabeça sem olhar a foto na caixa.
Na GoGenetic, priorizamos a montagem de novo. Essa escolha estratégica permite que identifiquemos genes inéditos, ilhas de patogenicidade ou operons metabólicos que poderiam ser perdidos em uma montagem por referência. Em microrganismos isolados de ambientes complexos ou com alto potencial biotecnológico, a montagem de novo é o padrão-ouro para garantir que a arquitetura genômica revelada seja a mais fiel possível à realidade biológica do seu isolado.
Para realizar essa tarefa com eficiência e precisão, o software não olha para a sequência inteira de uma vez. Ele divide cada leitura em subunidades menores e sobrepostas chamadas k-mers (onde “k” representa o comprimento dessa sequência, como 21, 33 ou 55 bases).
Imagine a frase: “PRECISÃO QUE CONECTA CIÊNCIA E FUTURO”. Se a quebrarmos em k-mers de 7 caracteres, teríamos “PRECISÔ, “RECISÃO”, “ECISÃO “, “CISÃO Q”, e assim por diante. O montador utiliza uma estrutura matemática chamada Grafo de De Bruijn: ele conecta esses k-mers que compartilham extremidades idênticas, criando um caminho lógico. É como se o software criasse uma malha rodoviária onde cada k-mer é um ponto de conexão; o caminho mais percorrido e coerente através dessa malha revela a sequência real e contínua do genoma.
Para esta missão, utilizamos o SPAdes, amplamente reconhecido como um dos montadores mais precisos e versáteis para genomas de microrganismos. O SPAdes se destaca por trabalhar com múltiplos tamanhos de k-mers simultaneamente, o que ajuda a resolver tanto regiões de baixa cobertura quanto regiões repetitivas complexas.
O resultado final desta etapa são os Contigs: sequências contínuas de DNA que representam grandes blocos do genoma. Quanto mais longos e menos numerosos forem os contigs, mais próxima da “perfeição” está a sua montagem.
Etapa 7: Anotação Funcional
Anotar um genoma significa identificar a localização exata dos genes e atribuir a eles uma função biológica provável. É como pegar o manual de instruções que acabamos de montar e colocar etiquetas em cada capítulo, parágrafo e frase, explicando o que cada um faz: “este gene produz uma enzima para digerir lactose”, “este aqui confere resistência a um antibiótico”, e assim por diante.
Nesta etapa, identificamos elementos essenciais como:
- CDS (Coding Sequences): Sequências que codificam proteínas.
- rRNA e tRNA: RNAs estruturais necessários para a síntese proteica.
A diferença entre procariotos e eucariotos
O processo de anotação difere significativamente dependendo do seu organismo de interesse:
- Bactérias: São mais diretas. Os genes são contínuos e densamente compactados. Para este grupo, utilizamos o Prokka, um pipeline extremamente rápido e preciso que coordena diversas ferramentas para identificar e anotar genes bacterianos de forma robusta.
- Fungos: São organismos mais complexos (eucariotos). Seus genes são interrompidos por sequências não codificantes chamadas íntrons. Para lidar com essa complexidade, utilizamos o Funannotate, que utiliza algoritmos avançados para prever a estrutura correta dos genes e integrar evidências de proteínas conhecidas para uma anotação de alta qualidade.
Output da anotação:
Ao final desta etapa, temos alguns arquivos estruturados que seguem o padrão internacional das grandes bases de dados (como o NCBI), sendo os principais deles:
.fna (Fasta Nucleic Acid): O arquivo das sequências de DNA montadas.
.faa (Fasta Amino Acid): As sequências de aminoácidos de todas as proteínas preditas.
.gff3 (General Feature Format): O “mapa” do genoma, contendo as coordenadas e descrições de cada gene.
A partir destes arquivos, abrem-se as portas para análises mais avançadas. É possível calcular a densidade gênica do organismo ou buscar manualmente genes de interesse específico para sua linha de pesquisa.
Além disso, esses arquivos são o dado de entrada para softwares especializados que exploramos conforme a necessidade do seu projeto, como o antiSMASH (para descoberta de clusters biossintéticos de metabólitos secundários), o KofamScan (para reconstrução de vias metabólicas completas) e o Abricate (para busca de genes de resistência e virulência). O genoma completo não é apenas um resultado, ele é uma base de dados própria para futuras descobertas.
Etapa 8: Classificação Comparativa (FastANI)
Ter o genoma anotado é excelente, mas onde esse organismo se encaixa na árvore da vida?
Embora o sequenciamento de genes marcadores como 16S rRNA ou ITS seja o ponto de partida, ele muitas vezes não tem resolução suficiente para distinguir espécies próximas. Para essa classificação, utilizamos o ANI (Average Nucleotide Identity – Identidade Nucleotídica Média).
O ANI é uma métrica que calcula a identidade média de todas as regiões compartilhadas entre dois genomas. Em vez de compararmos apenas um gene marcador, comparamos o conteúdo genômico global.
- O Limiar da Espécie: Na comunidade científica, o ponto de corte amplamente aceito é de 95%. Se o genoma do seu microrganismo apresenta um ANI ≥ 95% em relação a uma linhagem de referência, eles são considerados membros da mesma espécie.
Na GoGenetic, utilizamos a ferramenta FastANI para comparar o seu genoma contra os genomas de referência mais próximos depositados em bancos de dados globais. O resultado dessa análise é apresentado em um Clustermap: um mapa de calor que agrupa os genomas por similaridade, acompanhado de um dendrograma. Este diagrama visual facilita a compreensão da distância evolutiva e confirma a identidade taxonômica do seu isolado.

E se o meu genoma não atingir os 95% de ANI?
Caso o seu genoma apresente um valor de ANI consistentemente abaixo de 95% em relação a todas as espécies conhecidas no banco de dados, isso é um forte indicativo que você pode estar diante de uma nova espécie ainda não descrita.
Nesses casos, a análise de genoma completo torna-se a prova central para a publicação de uma nova linhagem, permitindo que você descreva não apenas o nome do novo organismo, mas todo o arsenal metabólico exclusivo que o diferencia de seus “parentes” mais próximos.
O que você recebe: o relatório completo
A entrega da GoGenetic é desenhada para que você possa transitar rapidamente do dado bruto à interpretação científica. Nosso relatório é intuitivo, mas preserva todo o rigor necessário para publicações em periódicos de alto impacto ou depósitos de patentes.
O pacote padrão de entrega inclui:
- Dados Brutos: Acesso integral às reads de sequenciamento (arquivos FASTQ).
- Genoma Montado: O arquivo final do seu microrganismo (FASTA).
- Estatísticas da Montagem: Tamanho total do genoma, número de contigs cobertura média estimada, etc.
- Arquivos de Anotação funcional do seu genoma
- Identificação baseada em 16S/ITS (validação)
- ANI com espécies relacionadas
- Posicionamento taxonômico preciso
- Clustermap com dendograma representando as relações filogenéticas com genomas próximos.
Análises personalizadas sob demanda: Para projetos que exigem um mergulho profundo na biologia sistêmica, oferecemos módulos adicionais:
- Mapeamento de Vias KEGG: Visualização de como os genes se organizam em rotas metabólicas.
- Identificação de vias completas, incompletas ou ausentes do seu interesse.
- Bioprospecção Direcionada: Busca por genes de interesse específico, como quitinases, fatores de virulência, resistência a antibióticos, etc.
- Sua demanda, nosso pipeline: Se a sua pesquisa exige uma métrica específica, uma comparação com bancos de dados proprietários ou uma análise customizada, você pode trazer a sua demanda diretamente para o nosso time técnico. Nós avaliamos a viabilidade científica e desenvolvemos uma análise personalizada para atender exatamente ao objetivo do seu projeto.
Especialização em Bioinsumos: do DNA ao Campo
Para a indústria de bioinsumos, desenvolvemos um Relatório Personalizado que vai muito além da taxonomia. Entendemos que o valor de um microrganismo promotor de crescimento ou agente de biocontrole está no seu arsenal bioquímico. Por isso, integramos análises de vias metabólicas e clusters de genes biossintéticos (BGCs) para entregar:
- Produção de Ácidos Orgânicos: Identificação do potencial de síntese de ácidos acético, glucônico, lático, málico e oxálico.
- Dinâmica de Fósforo: Mapeamento de genes de transporte e mecanismos de assimilação de fosfatos.
- Modulação Hormonal: Busca por vias de fitormônios essenciais, como auxinas, giberelinas, ácido salicílico, jasmonato e ácido abscísico, além da capacidade de degradação de etileno (ACC deaminase).
- Arsenal Enzimático e Antagonismo: Identificação detalhada de proteases, quitinases e metabólitos secundários, incluindo sideróforos, terpenos, antibacterianos, fungicidas, surfactantes e inseticidas.
- Ciclo do Nitrogênio: Análise completa das vias de fixação, nitrificação e desnitrificação.
- Inteligência em Bacillus: Para este gênero, realizamos uma busca dedicada por toxinas, como os genes cry, responsáveis pela produção de proteínas cristalinas com potente atividade inseticida, fundamentais para o desenvolvimento de biopesticidas.
Esses arquivos permitem:
- Realizar análises customizadas
- Comparar com outros genomas do seu laboratório
- Submeter a NCBI/EBI para número de acesso
- Publicar resultados em artigos científicos
O que a análise de Genoma completo pode revelar
1. Capacidades Metabólicas Completas
Diferente de testes bioquímicos tradicionais que avaliam um fenótipo ou uma enzima por vez, o genoma revela todo o potencial metabólico codificado no DNA do organismo.
Exemplo Real: Análise de um Bioinsumo.
Imagine que você isolou uma linhagem promissora para o setor agrícola. A análise de genoma completo revela:
- Identidade Taxonômica: confirmada como Priestia megaterium (anteriormente Bacillus megaterium) através de ANI.
- Acidificação do Meio: presença de rotas completas para produção dos ácidos orgânicos acético, glucônico, lático e málico, fundamentais para a solubilização de nutrientes no solo.
- Eficiência Nutricional: foram identificados 49 genes envolvidos especificamente no transporte e na assimilação de fosfatos, garantindo alta performance em solos com baixa disponibilidade de P.
- Biofábrica de Metabólitos: o genoma revelou clusters de biossíntese para sideróforos (sequestro de ferro), 3 tipos de terpenos e 2 compostos antibacterianos, conferindo potencial de biocontrole.
- Ciclo do Nitrogênio: capacidade genética de assimilar nitrito como fonte de nitrogênio.
- Limitação Conhecida: a análise revelou a ausência de genes para a produção de fitormônios.
Implicação: com esses dados, você não apenas conhece seu organismo, mas pode posicioná-lo estrategicamente no mercado. Você sabe que ele é um excelente solubilizador de fósforo e agente de biocontrole via sideróforos, mas que seu mecanismo de ação não passa pela via hormonal. Isso direciona o manejo, evita promessas técnicas equivocadas e acelera o registro do produto.
4. Potencial Biossintético
Ferramentas como antiSMASH analisam genomas em busca de clusters de genes biossintéticos como:
- Policetídeos (muitos antibióticos)
- Peptídeos não-ribossomais (penicilina, ciclosporina)
- Terpenos
- Lantipeptídeos
- Sideróforos
Descoberta surpreendente: Muitos genomas contêm 10-30 clusters biossintéticos, mas a maioria está silenciosa em condições de laboratório. O genoma revela potencial não expresso.
5. Comparação com patógenos ou organismos regulados
Segurança em aplicações: se você pretende usar o microrganismo comercialmente, o genoma pode:
- Confirmar ausência de genes de virulência
- Verificar ausência de toxinas
- Dar evidências de que o organismo não oferece risco
- Facilitar aprovação regulatória
6. Identificação de Novas Espécies
Se ANI <95% com todas as espécies conhecidas, você tem forte evidência de nova espécie.
Valor acadêmico:
- Descrição de nova espécie é publicável
- Você nomeia a espécie (eternizada na ciência) Citações garantidas por décadas
Por que fazer análise de Genoma completo?
Para pesquisa básica
Compreensão fundamental:
- Como esse organismo funciona em nível molecular?
- Quais são todas as suas capacidades?
- Como ele se relaciona evolutivamente com outros?
- Que adaptações únicas ele possui?
Para pesquisa aplicada
Desenvolvimento de aplicações:
- Quais genes são responsáveis pela atividade de interesse?
- Conhecer a estrutura gênica de um gene de interesse, facilitando estudos de engenharia genética visando um aumento de expressão da proteína.Existem outros genes que podem ser explorados?
- Como esse organismo se compara com alternativas comerciais?
Para Propriedade Intelectual
Proteção de descobertas:
- Sequências únicas servem como assinatura molecular
- Base para desenvolvimento de produtos comerciais
Para publicação científica
Requisito crescente:
- Muitos periódicos exigem genoma completo (não apenas 16S)
- Genomas aumentam impacto e citações de artigos
- Possibilitam análises comparativas por outros pesquisadores
- Bancos de dados públicos aumentam visibilidade
Quanto tempo leva?
4-5 semanas desde o recebimento da amostra até entrega do relatório padrão completo.
Compare com métodos anteriores (Sanger sequencing de genoma completo): 6-12 meses e custos 100x maiores.
Conclusão: o Genoma como ferramenta de descoberta
A análise de genoma completo transformou microbiologia de uma ciência descritiva em uma ciência preditiva e explicativa.
Você não precisa mais testar centenas de condições para descobrir o que seu microrganismo pode fazer. O genoma prediz capacidades, explica fenótipos observados, e revela potenciais muitas vezes não expressos.
É como a diferença entre tentar entender um carro apenas dirigindo (pesquisa tradicional) versus ter acesso ao manual de engenharia completo com todos os diagramas (genômica). Você ainda precisa dirigir para validar, mas sabe exatamente onde olhar e o que esperar.
Para o pesquisador moderno, a pergunta não é “devo sequenciar o genoma?”
A pergunta é: “que descobertas estou perdendo por não ter sequenciado ainda?”
- Contato: (41) 3024-0660 | (41) 99215-1776
- Email: comercial@gogenetic.com.br
- Redes sociais: @gogenetic | @gogeneticagro | @gosolos
Sobre a GoGenetic: Oferecemos serviços completos de sequenciamento e análise de genomas de bactérias, fungos e algas. Utilizamos tecnologia Illumina NextSeq com cobertura >30x, ferramentas bioinformáticas avançadas (SPAdes, Prokka, FastANI) e entregamos relatórios detalhados com montagem, anotação funcional e classificação comparativa. Entre em contato para discutir seu projeto de pesquisa.
Palavras-chave: análise de genoma completo, sequenciamento de genoma bacteriano, sequenciamento de genoma fúngico, genoma de algas, Illumina NextSeq, SPAdes, Prokka, FastANI, ANI, anotação genômica, montagem de genoma, sequenciamento NGS, genoma microbiano, análise bioinformática, identificação molecular microrganismos
Referências Científicas
[1] Koonin, E.V. & Wolf, Y.I. (2008). Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Research, 36(21), 6688-6719. https://doi.org/10.1093/nar/gkn668
[2] Mohanta, T.K. & Bae, H. (2015). The diversity of fungal genome. Biological Procedures Online, 17, 8. https://doi.org/10.1186/s12575-015-0020-z
[3] Blanc, G. et al. (2010). The Chlorella variabilis NC64A genome reveals adaptation to photosymbiosis, coevolution with viruses, and cryptic sex. The Plant Cell, 22(9), 2943-2955. https://doi.org/10.1105/tpc.110.076406
[4] Kuo, C.H. & Ochman, H. (2009). Deletional bias across the three domains of life. Genome Biology and Evolution, 1, 145-152. https://doi.org/10.1093/gbe/evp016
[5] Sims, D. et al. (2014). Sequencing depth and coverage: key considerations in genomic analyses. Nature Reviews Genetics, 15(2), 121-132. https://doi.org/10.1038/nrg3642
[6] Sambrook, J. & Russell, D.W. (2001). Molecular Cloning: A Laboratory Manual, 3rd Edition. Cold Spring Harbor Laboratory Press.
[7] Schoch, C.L. et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proceedings of the National Academy of Sciences, 109(16), 6241-6246. https://doi.org/10.1073/pnas.1117018109
[8] Adey, A. et al. (2010). Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biology, 11(12), R119. https://doi.org/10.1186/gb-2010-11-12-r119
[9] Illumina (2020). An Introduction to Next-Generation Sequencing Technology. Technical Note: Sequencing.
[10] Bentley, D.R. et al. (2008). Accurate whole human genome sequencing using reversible terminator chemistry. Nature, 456(7218), 53-59. https://doi.org/10.1038/nature07517
[11] Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Babraham Bioinformatics. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
[12] Bolger, A.M. et al. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114-2120. https://doi.org/10.1093/bioinformatics/btu170
[13] Bankevich, A. et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology, 19(5), 455-477. https://doi.org/10.1089/cmb.2012.0021
[14] Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics, 30(14), 2068-2069. https://doi.org/10.1093/bioinformatics/btu153
[15] Jain, C. et al. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nature Communications, 9, 5114. https://doi.org/10.1038/s41467-018-07641-9
[16] Richter, M. & Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proceedings of the National Academy of Sciences, 106(45), 19126-19131. https://doi.org/10.1073/pnas.0906412106
[17] Galperin, M.Y. & Koonin, E.V. (2010). From complete genome sequence to ‘complete’ understanding? Trends in Biotechnology, 28(8), 398-406. https://doi.org/10.1016/j.tibtech.2010.05.006
[18] Blin, K. et al. (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Research, 47(W1), W81-W87. https://doi.org/10.1093/nar/gkz310
[19] Doroghazi, J.R. & Metcalf, W.W. (2013). Comparative genomics of actinomycetes with a focus on natural product biosynthetic genes. BMC Genomics, 14, 611. https://doi.org/10.1186/1471-2164-14-611



